De Bits a Biología #1 Utilizando el algoritmo LCS para el alineamiento global de secuencias en Biología Computacional
Bits to Biology #1 Using the LCS algorithm for global sequence alignment in Computational Biology
Un análisis paso a paso del problema de la Subsecuencia Común Más Larga y el problema de alineación de secuencias con una explicación de sus soluciones destacando sus similitudes y diferencias
Introducción
¡Bienvenido! Has encontrado la primera entrada de mi serie “De Bits a Biología”, donde exploro las conexiones entre algoritmos generales que puedes encontrar en una clase de ciencias de la computación y la biología computacional/bioinformática para hacerlos más intuitivos y accesibles para aquellos sin formación formal en biología. Me tomó algún tiempo convencerme de que no necesariamente necesitas un conocimiento formal en biología para hacer contribuciones significativas en biología computacional, un campo increíblemente fascinante, y espero convencerte de que lo intentes si estás en una situación similar 🙂
En este artículo, estamos revisando la Alineación de Secuencias, uno de los problemas más fundamentales en biología computacional. La alineación de secuencias de ADN, ARN y proteínas tiene una variedad de implicaciones profundas, incluyendo la comprensión de las relaciones evolutivas, el avance de la anotación funcional, la identificación de mutaciones y el diseño de medicamentos y la medicina de precisión.
Tabla de contenidos
- Subsecuencia Común Más Larga (LCS) – Descripción general de LCS – Solución de programación dinámica de LCS – Implementación en Python de LCS
- Alineación Global de Secuencias (GSA) – Descripción general de GSA – Sistemas de puntuación – Solución de programación dinámica de GSA – Implementación en Python de GSA
- LCS vs. Alineación Global de Secuencias: Similitudes y diferencias – Tabla de resumen
Problema de la Subsecuencia Común Más Larga (LCS)
La Subsecuencia Común Más Larga (LCS) es un problema clásico de ciencias de la computación que identifica la subsecuencia más larga entre todas las subsecuencias válidas compartidas entre 2 o más secuencias de entrada.
Aclararemos la definición del problema LCS al notar primero una distinción importante entre una subcadena y una subsecuencia. Toma la secuencia "ABCDE":
- Desmitificando el Aprendizaje Automático Bibliotecas y Herramientas Populares de ML
- 5 funciones sencillas de Python que puedes empezar a utilizar hoy para escribir un código mejor
- 3 consejos útiles de Pandas para trabajar con datos de fecha y hora
- Una subcadena consiste en caracteres contiguos en su orden original. Por ejemplo, subcadenas válidas incluyen
"ABC",“CD”, etc., pero no“ABDE”o“CBA”. - Una subsecuencia, por otro lado, no tiene que ser contigua pero también debe estar en el orden original. Por ejemplo, subsecuencias válidas incluyen
“AC”y“BFG”.
La LCS, entonces, es la más larga entre todas las posibles subsecuencias entre 2 o más secuencias. Aquí tienes un ejemplo rápido:

En la práctica, LCS tiene muchas aplicaciones que involucran datos de texto mucho más grandes, como la detección de plagio¹ o el comando diff de Unix, lo que requiere una solución algorítmica eficiente y confiable y escalable.
Formalizando la solución de LCS
Aquí tienes un repaso de la solución de programación dinámica de 3 pasos para LCS:
- Inicialización (crear una matriz m × n, donde m y n son las longitudes de la secuencia 1 y las secuencias 2).
- Rellenar la matriz según la fórmula de recurrencia dada a continuación.
- Retroceder para encontrar la LCS y su longitud.
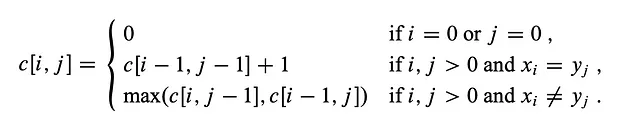
Teniendo en cuenta las entradas "LONGEST" y "STONE". Basados en la relación de recurrencia dada anteriormente, completamos la matriz DP y luego retrocedemos de la siguiente manera:
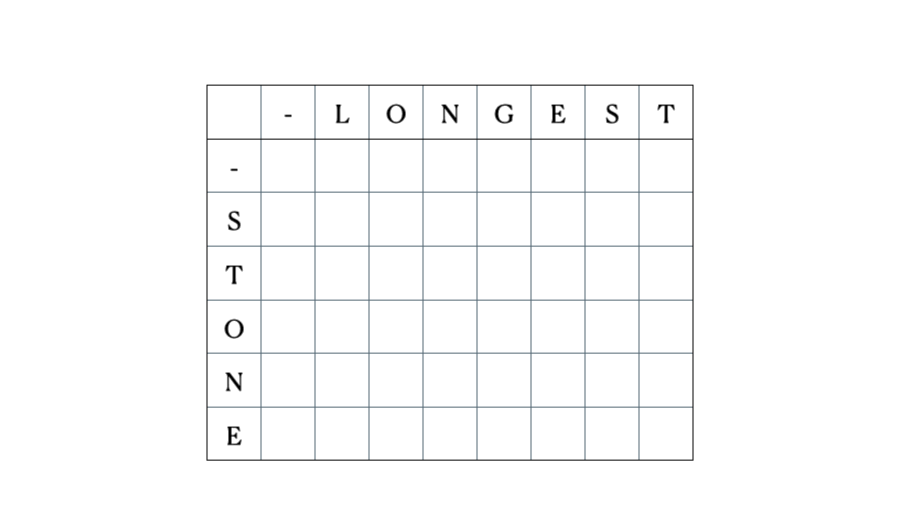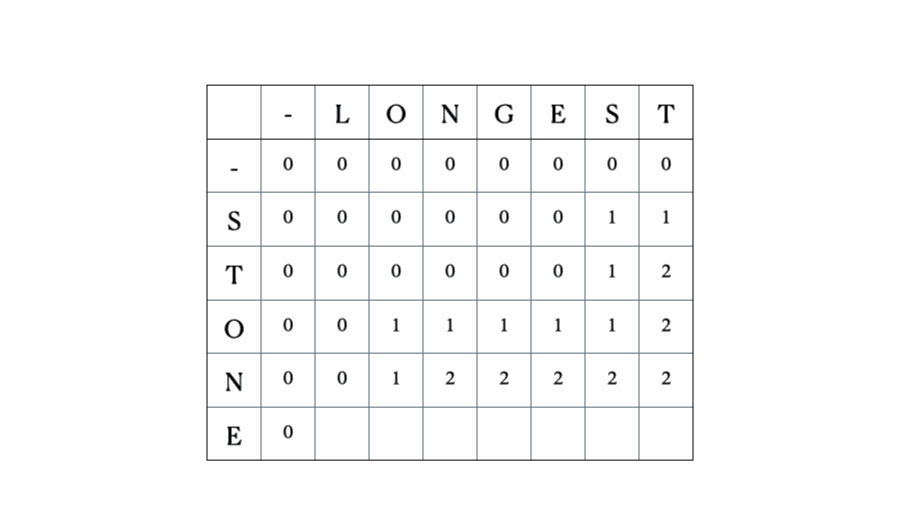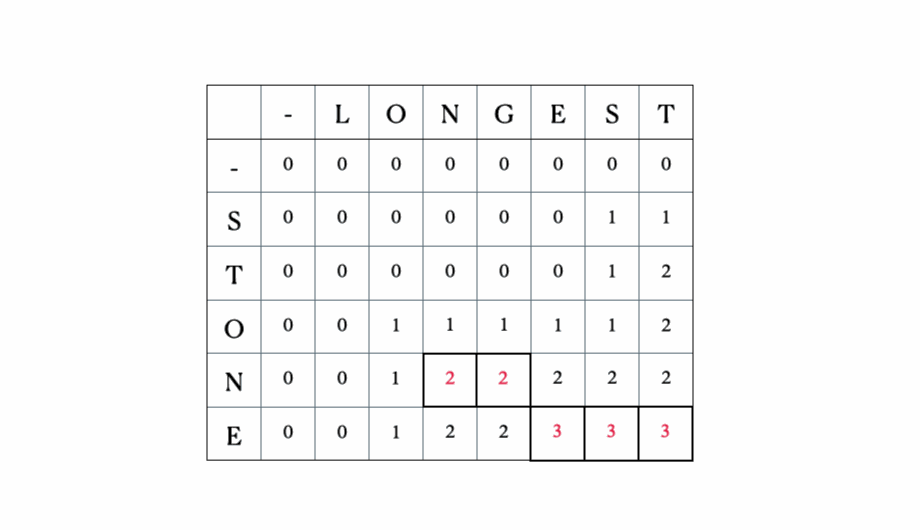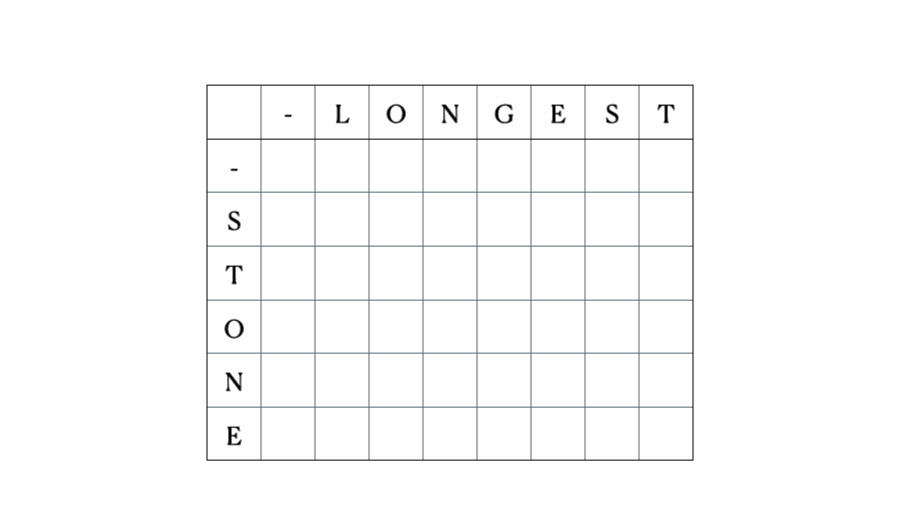
Esta es la tabla DP completa:
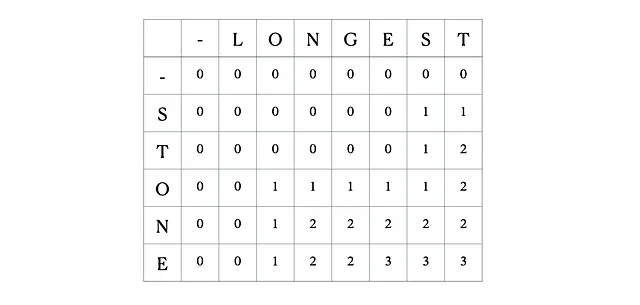
Esto se traduce en la implementación de Python a continuación:
# Longest Common Subsequence
def lcs(seq1, seq2):
m = len(seq1)
n = len(seq2)
# Construir tabla dp
table = [[0 for x in range(n+1)] for x in range(m+1)]
# Rellenar tabla basada en fórmula recursiva (15.9) de CLRS
for i in range(m+1):
for j in range(n+1):
if i == 0 or j == 0: # caso 1
table[i][j] = 0
elif seq1[i-1] == seq2[j-1]: # caso 2
table[i][j] = table[i-1][j-1] + 1
else: # caso 3
table[i][j] = max(table[i-1][j], table[i][j-1])
# Comenzar retroceso desde la celda inferior derecha
index = table[m][n]
lcs = [""] * (index+1)
lcs[index] = ""
i = m
j = n
while i > 0 and j > 0:
if seq1[i-1] == seq2[j-1]: # agregar caracteres coincidentes a LCS
lcs[index-1] = seq1[i-1]
i -= 1
j -= 1
index -= 1
elif table[i-1][j] > table[i][j-1]:
i -= 1
else:
j -= 1
# Imprimir la subsecuencia común más larga y su longitud
print("Secuencia 1: " + seq1 + "\nSecuencia 2: " + seq2)
print("LCS: " + lcs + ", longitud: " + str(len(lcs)))Ejemplo de uso:
seq1 = 'STONE'
seq2 = 'LONGEST'
lcs(seq1, seq2)Salida:
Secuencia 1: STONE
Secuencia 2: LONGEST
LCS: ONE, longitud: 3Ahora, veamos cómo se utiliza este algoritmo en… ↓
Problema de alineación de secuencias (global)
¿Por qué queremos alinear secuencias de ADN/ARN/proteínas?
En términos intuitivos, alinear dos secuencias es una tarea de localizar segmentos idénticos entre las dos secuencias, sirviendo el objetivo final de evaluar el nivel de similitud entre ellas. Relativamente sencilla en concepto, esta tarea siempre ha sido bastante compleja en la práctica debido a los innumerables cambios que pueden ocurrir en las secuencias de ADN y proteínas a través de la evolución. Específicamente, ejemplos comunes de estos cambios incluyen:
- Mutación puntual, a través de inserción, eliminación o sustitución: – Ejemplo de inserción: AAA → ACAA – Ejemplo de eliminación: AAA → AA – Ejemplo de sustitución: AAA → AGA
- Fusión de secuencias o segmentos de diferentes genes
Estos cambios no deseados pueden introducir ambigüedades y diferencias que ocultan nuestra comprensión de una determinada secuencia. Es aquí donde utilizamos técnicas de alineación para evaluar la similitud entre secuencias e inferir características como homología.
¿Cómo alineamos secuencias?
Debido a estos cambios evolutivos y a la naturaleza de las comparaciones de secuencias biológicas, a diferencia de en LCS donde simplemente descartamos los elementos “no coincidentes” y solo mantenemos el segmento común, en la alineación global de secuencias debemos alinear todos los caracteres, lo que significa que las alineaciones a menudo involucran 3 componentes:
- Coincidencias, indicadas por
|entre las bases coincidentes - No coincidencias, indicadas por espacios en blanco entre las bases que no coinciden
- Inserciones de espacios, indicadas por guiones
—
Aquí hay un ejemplo. Supongamos que tenemos dos secuencias de ADN “ACCCTG” y “ACCTGC”. Hay varias formas de hacer coincidirlas base a base, teniendo en cuenta las 3 operaciones de las que hablamos antes:
Podríamos alinearlas sin ninguna inserción de espacio:
ACCCTGT||| |ACCTGCTEsta alineación tiene 0 inserciones de espacio, 3 coincidencias, seguidas de 3 no coincidencias, y luego 1 coincidencia al final.
O podríamos permitir inserciones de espacio a cambio de más coincidencias:
ACCCTG-T||| || |ACC-TGCTEsta alineación tiene 6 coincidencias totales, 2 espacios y 0 no coincidencias (no contamos un par de base-espacio como una no coincidencia, ya que ya se penaliza con la inserción del espacio).
O, si realmente quisiéramos, también podríamos alinearlas de esta manera, lo cual resultaría en solo 1 coincidencia total:
-ACC-CT-GT | ACCTG-CT--Entre cualquier par de secuencias, casi siempre hay infinitas formas de generar alineaciones técnicamente correctas como acabamos de ver. Algunas de ellas serán claramente peores que otras, pero habrá algunas que sean “aproximadamente igual de buenas”, como las opciones 1 y 2 anteriores.
Entonces, ¿cómo evaluamos la calidad de cada alineación, elegimos la alineación óptima y resolvemos empates si es necesario?
Esquemas de puntuación
1. ACCCTGT 2. ACCCTG-T 3. -ACC-CTGT ||| | ||| || | | ACCTGCT ACC-TGCT ACCTG-CT-En el ejemplo que vimos anteriormente, cada alineación involucra una combinación diferente de números de coincidencias, no coincidencias y espacios.
A diferencia del problema de Subsecuencia Común más Larga (LCS, por sus siglas en inglés) donde una subsecuencia solo se evalúa por su longitud — cuanto más larga, mejor —, en la alineación de secuencias, a menudo medimos la calidad de una alineación definiendo un sistema de puntuación cuantitativo que abarca los 3 componentes clave de cada emparejamiento base a base:
- Recompensa por coincidencia: la puntuación agregada a la puntuación total de la alineación cada vez que hay un par de bases coincidentes en nuestras secuencias.
- Penalización por espacio: el valor de penalización que se resta de la puntuación total de la alineación cada vez que introducimos un espacio.
- Penalización por no coincidencia: el valor de penalización que se resta de la puntuación total de la alineación cada vez que tenemos una no coincidencia.
¿Cómo formalizamos esto como un algoritmo?
La solución algorítmica se superpone significativamente con la solución del problema LCS en términos de estructura. También es un proceso de 3 pasos, conocido como el algoritmo Needleman-Wunsch:
- Inicialización (crear una matriz m × n, donde m y n son las longitudes de la secuencia 1 y las secuencias 2)
- Rellenar la matriz según la fórmula recursiva dada a continuación
- Retroceso para encontrar la alineación óptima y su longitud
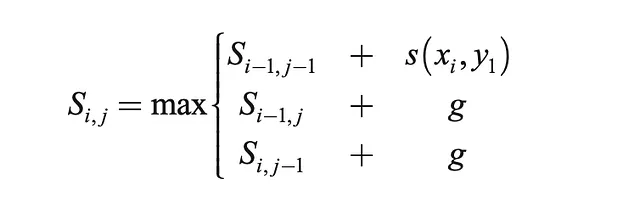
Antes de llegar a la formulación matemática y en Python de la solución, aquí hay una ilustración rápida del proceso con las secuencias “ACGTGTCAG” y “ATGCTAG”:
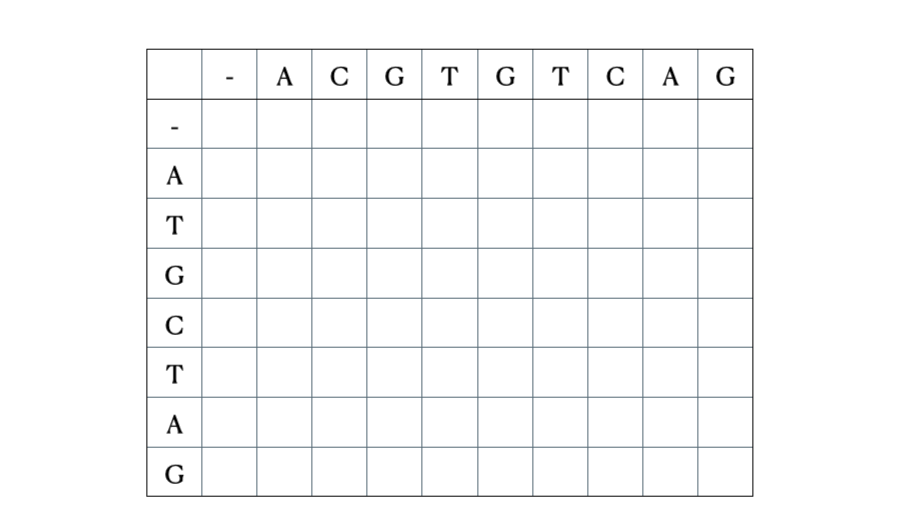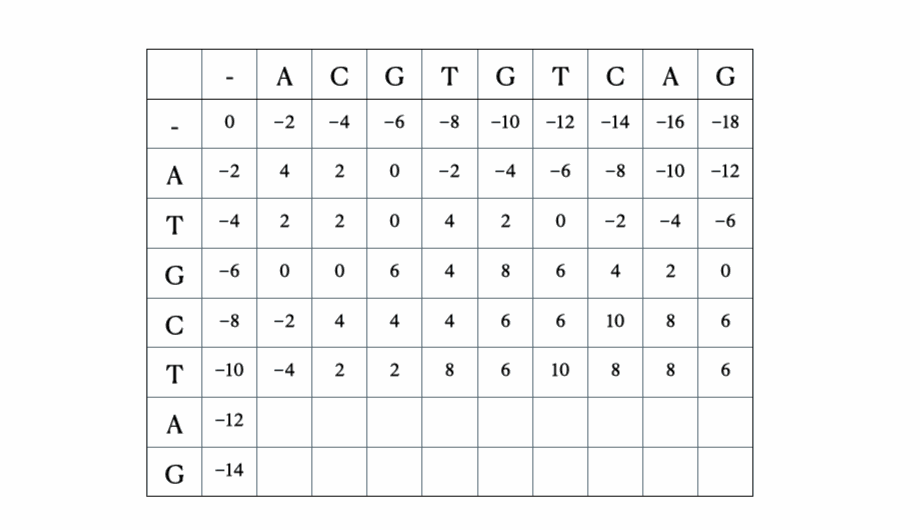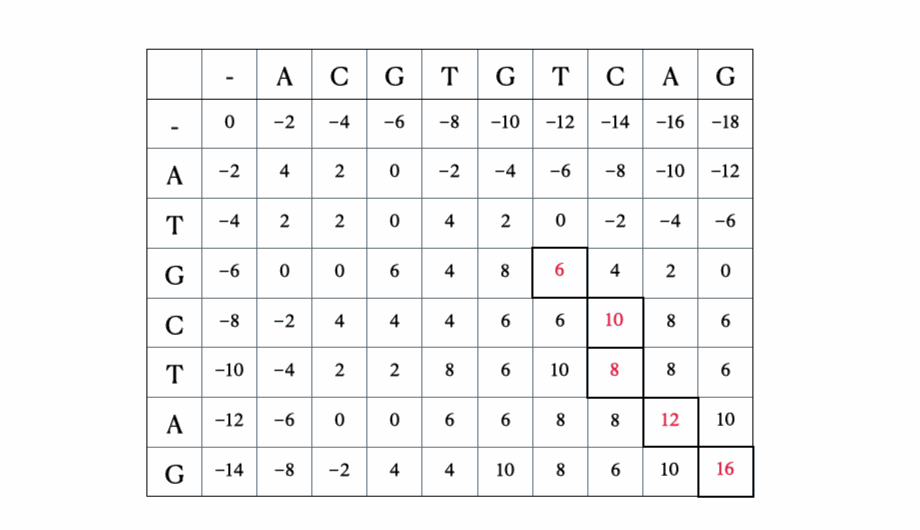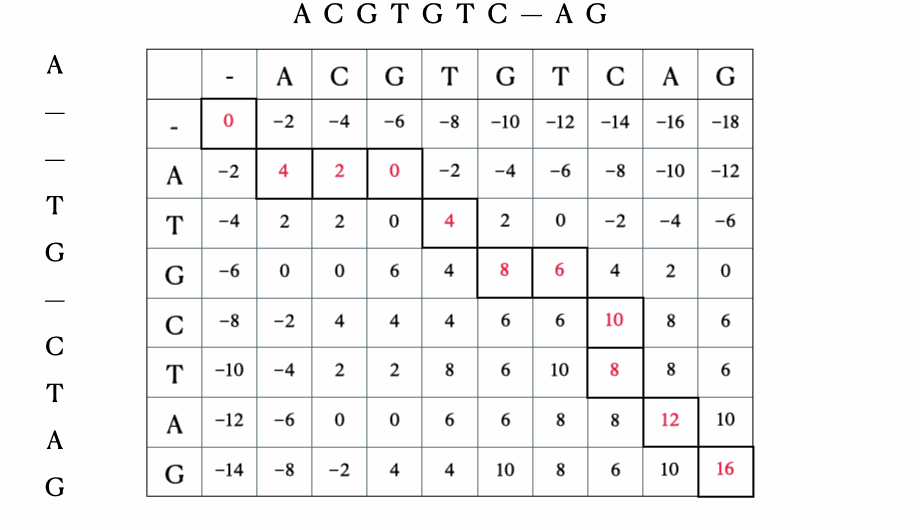
Solución en Python
Vamos a implementar la solución en Python utilizando una clase, GlobalSeqAlign, que se encarga tanto de completar la matriz como del traceback.
Primero, construimos un objeto que contiene las 5 piezas de información clave: secuencia 1, secuencia 2, puntaje de coincidencia, penalización por desajuste y penalización por brecha.
class globalSeqAlign: def __init__(self, s1, s2, M, m, g): self.s1 = s1 self.s2 = s2 self.M = M self.m = m self.g = gLuego, definimos una función auxiliar que obtiene el puntaje entre dos bases dependiendo de si son una coincidencia, desajuste o pareja de bases y brecha:
def base_pair_score(self, char1, char2): if char1 == char2: return self.M elif char1 == '-' or char2 == '-': return self.g else: return self.mAhora, definimos una función muy similar a la función LCS que vimos anteriormente, que utiliza una matriz m × n, la completa según la fórmula recursiva y luego realiza el traceback para devolver el alineamiento óptimo que maximiza el puntaje total:
def optimal_alignment(self, s1, s2): m = len(s1) n = len(s2) score = [[0 for x in range(n+1)] for x in range(m+1)] # Calcular tabla de puntajes for i in range(m+1): # Inicializar con 0s score[i][0] = self.g * i for j in range(n+1): # Inicializar con 0s score[0][j] = self.g * j for i in range(1, m+1): # Completar celdas restantes según la fórmula recursiva anterior for j in range(1, n+1): match = score[i-1][j-1] + self.base_pair_score(s1[j-1], s2[i-1]) delete = score[i-1][j] + self.g insert = score[i][j-1] + self.g score[i][j] = max(match, delete, insert) # Traceback, comenzando desde la celda inferior derecha align1 = "" align2 = "" i = m j = n while i > 0 and j > 0: score_curr = score[i][j] score_diag = score[i-1][j-1] score_up = score[i][j-1] score_left = score[i-1][j] if score_curr == score_diag + self.base_pair_score(s1[j-1], s2[i-1]): # coincidencia align1 += s1[j-1] align2 += s2[i-1] i -= 1 j -= 1 elif score_curr == score_up + self.g: # hacia arriba align1 += s1[j-1] align2 += '-' j -= 1 elif score_curr == score_left + self.g: # hacia la izquierda align1 += '-' align2 += s2[i-1] i -= 1 while j > 0: align1 += s1[j-1] align2 += '-' j -= 1 while i > 0: align1 += '-' align2 += s2[i-1] i -= 1 return(align1[::-1], align2[::-1]) # Orden inversoUna vez que tenemos el alineamiento óptimo, podemos proceder con el análisis cuantificando qué tan bien coinciden las secuencias.
Cuantificación de la similitud: similitud porcentual
Una de las formas más simples de medir el “nivel de similitud” es a través de la similitud porcentual. Tomamos el alineamiento óptimo, observamos su longitud y la dividimos por la longitud total de las regiones coincidentes.
Por ejemplo:
Seq 1: ACACAGTCAT |||| |||||Seq 2: ACACTGTCATHay 9 coincidencias de las 10 parejas totales, lo que da como resultado una similitud del 90%. Hay muchas otras formas interesantes de analizar la calidad de un alineamiento, pero el mecanismo subyacente de alineamiento de secuencias sigue siendo el mismo.
Comparación de LCS y Alineamiento Global de Secuencias
Ahora que hemos analizado detalladamente tanto los problemas como sus soluciones, veamos el LCS (Subsecuencia Común más Larga) y el alineamiento global de secuencias lado a lado.
Similitudes
- Objetivo: ambos problemas encuentran y maximizan las similitudes y elementos comunes en las secuencias de entrada.
- Enfoque algorítmico: ambos problemas pueden resolverse utilizando programación dinámica, lo cual implica una matriz/tabla de PD, completar la matriz y retroceder para producir las secuencias/alineamientos.
- Esquema de puntuación: ambos problemas tienen puntuaciones asociadas con coincidencias, no coincidencias (y huecos) en la coincidencia de secuencias. Estas puntuaciones se utilizan explícita e implícitamente en el algoritmo para obtener la subsecuencia/alineamiento óptimo/a.
Diferencias
- Sistemas de puntuación: LCS adopta automáticamente un esquema de puntuación de puntuación de coincidencia = 1, puntuación de no coincidencia/hueco = 0; el alineamiento global de secuencias tiene un esquema de puntuación más complejo definido por puntuación de coincidencia = A, penalización por no coincidencia = B, penalización por hueco = C.
- Manejo de huecos: LCS generalmente trata los huecos como no coincidencias porque, según la definición de subsecuencias, solo conservamos las coincidencias; el alineamiento global de secuencias generalmente asigna una puntuación de penalización a los huecos, diferente de la penalización por no coincidencia, si hay una preferencia por la no coincidencia/hueco.
- Longitud del alineamiento: LCS se preocupa por encontrar un segmento común a todas las entradas, sin importar los elementos que no formen parte de este segmento; el alineamiento global de secuencias coincide con todas las secuencias dadas, incluso si eso significa insertar huecos si las secuencias de entrada difieren en longitud.
Aquí tienes una tabla resumen de los puntos anteriores:

Conclusión
¡Gracias por acompañarme hasta aquí 😼! En este artículo, nos hemos centrado en un problema específico de programación dinámica: la Subsecuencia Común más Larga (LCS), y hemos realizado una comparación lado a lado con su aplicación en la biología computacional para el Alineamiento de Secuencias.
Más adelante en esta serie, también analizaremos otros temas, como los Modelos Ocultos de Markov, los Modelos de Mezclas Gaussianas y los algoritmos de Clasificación, y cómo resuelven problemas específicos en la biología computacional. ¡Hasta pronto y feliz programación ✌🏼!
[1] Baba, K., Nakatoh, T., & Minami, T. (2017). Detección de plagio utilizando similitud de documentos basada en representación distribuida. Procedia Computer Science, 111, 382–387. https://doi.org/10.1016/j.procs.2017.06.038
[2] Cormen, T. H. (Ed.). (2009). Introducción a los algoritmos (3.ª ed.). MIT Press.
[3] Zvelebil, M. J., & Baum, J. O. (2008). Comprender la bioinformática. Garland Science.
We will continue to update Zepes; if you have any questions or suggestions, please contact us!
Was this article helpful?
93 out of 132 found this helpful
Related articles
- Procesamiento de datos a gran escala con MapReduce
- 10 Mejores Generadores de Presentaciones de IA
- Ciencia de Datos Acelerada Intérprete de Código ChatGPT como tu Asistente de IA
- Este documento de IA muestra cómo la toxicidad de ChatGPT puede aumentar hasta seis veces cuando se le asigna una personalidad
- Construyendo visualizaciones interactivas de datos en Python Una introducción a Plotly
- Cómo la IA Generativa puede apoyar a las empresas de la industria alimentaria
- Conoce a RoboPianist una nueva suite de referencia para el control de alta dimensionalidad en el dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del dominio del






